
使用 AutoDock Vina 进行分子对接
主要记录在 Ubantu 上使用 Vina 进行分子对接的过程。大致可分为受体配体分子的预处理(MGLTools)和分子对接(Vina)两部分。
1 MGLTools 和 Vina 的下载及安装
2.1 MGLTools 的下载及安装
wget http://mgltools.scripps.edu/downloads/downloads/tars/releases/REL1.5.6/mgltools_x86_64Linux2_1.5.6.tar.gz
tar -zxvf mgltools_x86_64Linux2_1.5.6.tar.gz
cd mgltools_x86_64Linux2_1.5.6
./install.sh
注意:
① 上方链接可能会因软件更新而失效,以官网(见参考资料)链接为准。
② install.sh 运行结束后会出现如下提示,就是让添加该软件到环境变量中,并且重定义4个指令,可以不进行这些设置,但是调用的时候需要使用全路径。
-- add the /data/yangpp/soft/mgltools_x86_64Linux2_1.5.6/bin to the path environment variable in .cshrc or .bashrc:
.cshrc:
set path = (/data/yangpp/soft/mgltools_x86_64Linux2_1.5.6/bin $path)
.bashrc
export PATH=/data/yangpp/soft/mgltools_x86_64Linux2_1.5.6/bin:$PATH
-- create aliases in your .cshrc or .bashrc
.cshrc:
alias pmv /data/yangpp/soft/mgltools_x86_64Linux2_1.5.6/bin/pmv
alias adt /data/yangpp/soft/mgltools_x86_64Linux2_1.5.6/bin/adt
alias vision /data/yangpp/soft/mgltools_x86_64Linux2_1.5.6/bin/vision
alias pythonsh /data/yangpp/soft/mgltools_x86_64Linux2_1.5.6/bin/pythonsh
.bashrc
alias pmv='/data/yangpp/soft/mgltools_x86_64Linux2_1.5.6/bin/pmv'
alias adt='/data/yangpp/soft/mgltools_x86_64Linux2_1.5.6/bin/adt'
alias vision='/data/yangpp/soft/mgltools_x86_64Linux2_1.5.6/bin/vision'
alias pythonsh='/data/yangpp/soft/mgltools_x86_64Linux2_1.5.6/bin/pythonsh'
-- source ./initMGLtools.sh (bash) or ./initMGLtools.csh (c-shell)
2.2 Vina 的下载及安装
wget http://vina.scripps.edu/download/autodock_vina_1_1_2_linux_x86.tgz
tar -zxvf autodock_vina_1_1_2_linux_x86.tgz
./autodock_vina_1_1_2_linux_x86/bin/vina --help
如需添加环境变量,在 ~/.bashrc 中添加下面内容后运行 source ~/.bashrc 即可。
export PATH="/data/yangpp/soft/autodock_vina_1_1_2_linux_x86/bin:$PATH"
2 受体配体分子的预处理
2.1 受体分子预处理
受体分子需要移除水分子,可以直接在 pdb 文件中找到并删除水分子的行,也可以在 pymol 中使用 remove waters 来移除水分子。之后就需要使用MGLTools 中的脚本将 pdb 文件转换成为 pdbqt 文件,脚本使用示例如下。
/data/yangpp/soft/mgltools_x86_64Linux2_1.5.6/bin/python /data/yangpp/soft/mgltools_x86_64Linux2_1.5.6/MGLToolsPckgs/AutoDockTools/Utilities24/prepare_receptor4.py -r receptor_protein.pdb
注意:
① 上面脚本看起来非常长,是因为没有将 MGLTools 添加到环境变量中,使用全路径调用脚本造成的。
② 提供的 python 脚本必须使用 MGLTools 自带的 python 来调用,因为现在应该没谁还有装 python2.5 吧 !
2.2 配体分子预处理
大致过程同上,将 pdb 文件转换为 pdbqt 文件,脚本如下。
/data/yangpp/soft/mgltools_x86_64Linux2_1.5.6/bin/python /data/yangpp/soft/mgltools_x86_64Linux2_1.5.6/MGLToolsPckgs/AutoDockTools/Utilities24/prepare_ligand4.py -l ligand_molecular.pdb
注意两个脚本后面所接的参数是不同的!!!
3 分子对接
3.1 分子对接
由于 vina 需要设置的参数相对较多,所以建议使用 config 文件来简化指令长度,此次使用的 config 文件内容如下。
ligand = ligand_molecular.pdbqt
receptor = receptor_protein.pdbqt
out = ligand_receptor_out.pdbqt
center_x = 66.172
center_y = 66.844
center_z = 79.808
size_x = 30
size_y = 30
size_z = 30
seed = 1234
直接运行 vina --config config.txt 来进行对接,一般数分钟即可完成对接。对接结束后会打印出如下图的提示并将所有的对接结果存放在参数 out 后填写的 pdbqt 文件中。
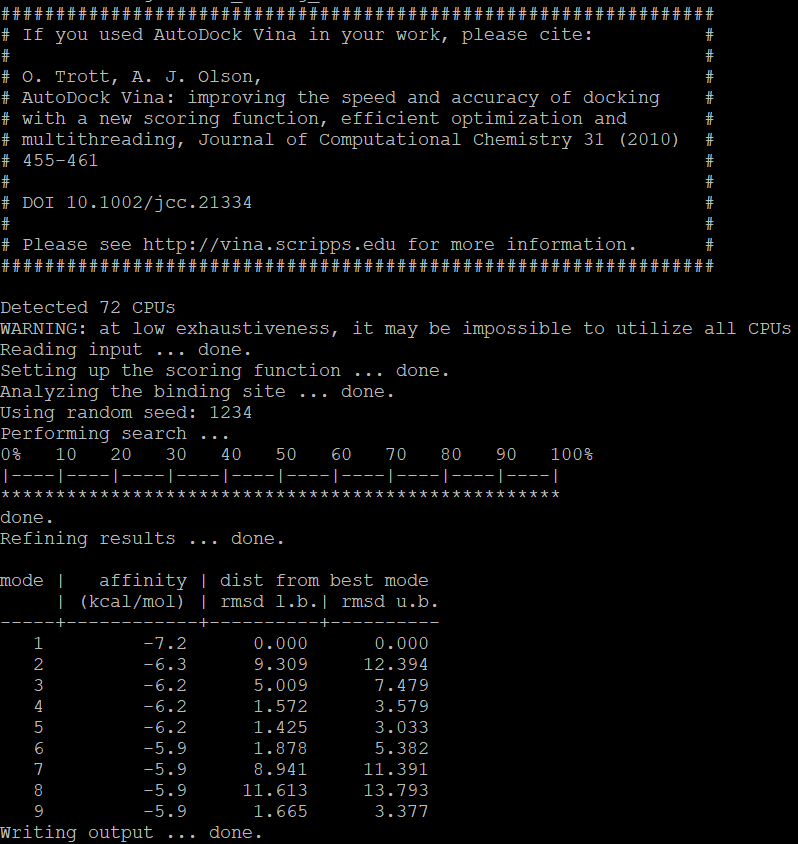
vina 中相关参数含义如下:
ligand, receptor and out: 配体、受体和输出的 pdbqt 文件。
center_x, center_y, center_z: 搜索空间的中心坐标。
size_x, size_y, size_z: 以中心坐标为源点的搜索空间范围,单位为埃。
seed: 随机种子,确保结果的可重复性。
log: 写出日志文件。
cpu: 设置使用的线程数。
num_modes: 最大计算的模型数目,默认计算出 9 个模型。
energy_range: 与最优结合模型相差的最大能量值(默认3),假如最优的能量值为-7.2 kcal/mol,那么最多计算到 -4.2 kcal/mol 的模型就会停止。
exhaustiveness: 用来控制对接的细致程度(默认8),与时间成正比。
看完上面参数含义,可以明显感觉到前三行的参数是必须填写的,其中搜索空间的中心坐标不能直接确定并且相对重要。所以中心坐标的可以通过如下几种方式来确定。
① 本次使用的是与配体分子结构较为类似的分子结合在受体上的大致中心位置。
② 使用 Protein-ligand binding site prediction 网页工具进行位置预测。
③ 使用 Discovery studio 软件进行预测(未亲测)。
3.2 对接结果的转换和查看
所有的对接结果都会存储在所设置的 pdbqt 格式文件中,并且其中只有配体分子的坐标信息,所以想查看整体的三维结构,首先需要将 pdbqt 格式再转换回 pdb 格式,再将该配体的 pdb 文件和受体的 pdb 文件合并到一个新的 pdb 文件中再使用 pymol 查看。
MGLTools 中提供了pdbqt_to_pdb.py 脚本来进行格式的转换,使用示例如下。
# pdbqt to pdb
/data/yangpp/soft/mgltools_x86_64Linux2_1.5.6/bin/python /data/yangpp/soft/mgltools_x86_64Linux2_1.5.6/MGLToolsPckgs/AutoDockTools/Utilities24/pdbqt_to_pdb.py -f ligand_receptor_out.pdbqt
# merge ligand receptor
cat receptor_protein.pdb ligand_receptor_out.pdbqt > complex.pdb
注意:
① 其中 complex.pdb 存放的是配体的 MODEL 1 和受体的结构,使用 pymol 查看即可。
② 为什么只有 MODEL 1 的结构?因为 pdbqt_to_pdb.py 只会转换第一个 MODEL,如果需要查看其它 MODEL 的三维结构,只需将其它 MODEL 的信息单独提取并转换即可。
参考资料:
1.MGLTools
2.MGLTools Downloads
3.AutoDock Vina
4.AutoDock Vina Downloads
5.教程:用AutoDock Vina进行分子对接
6.autodock vina官方视频教程
7.分子对接 Part1-Autodock 安装
8.分子对接 part2-蛋白与小分子3D结构前处理
9.分子对接 part3-蛋白与小分子对接
10.分子对接 part4-对接结果图像处理(PyMOL)
11.分子对接-问题答疑(1)/autogrid box选取
12.分子对接-问题答疑(2)/ PyMol可视化分析
13.分子对接-问题答疑(3)/pdb格式转换/对接出错
14.autodock-vina分子对接
15.autodock vina 从安装到分子对接