FastTree快速入门(Windows版)
以前接到过在 Windows 下使用 FastTree 的任务,由于当时理解能力还不够,并且网上关于 FastTree 的说明太少(可能是由于官方文档太详细了!),对于接触命令行的人来说可能比较好理解,但是对于纯小白来说,可能就会有点困难(反正我还是小白的时候理解起来比较困难),针对此情况就有写此文档的想法。
1.下载
进入 FastTree官方网站 ,在官网上找到下图所示界面,根据情况下载对应版本的软件,本文主要对 Windows 下 FastTree 的使用进行说明,所以本文下载的是 Windows 版本的 FastTree 软件。

下载完成后会出现 FastTree.exe 的文件,可放到任意文件夹中,但是必须知道此文件的路径。可用如下图方式查看文件的路径,右键此文件后点击最下方的“属性”,弹出对话框如下图,复制框中所示内容。

2.进入cmd界面
右键屏幕最左下方的 Windows键,出现下图点击 运行 (或使用键盘上的 Windows键 + R),输入 cmd 后回车即可进入命令界面。
注意:FastTree是免安装的软件,直接可用!!!
3.FastTree的使用
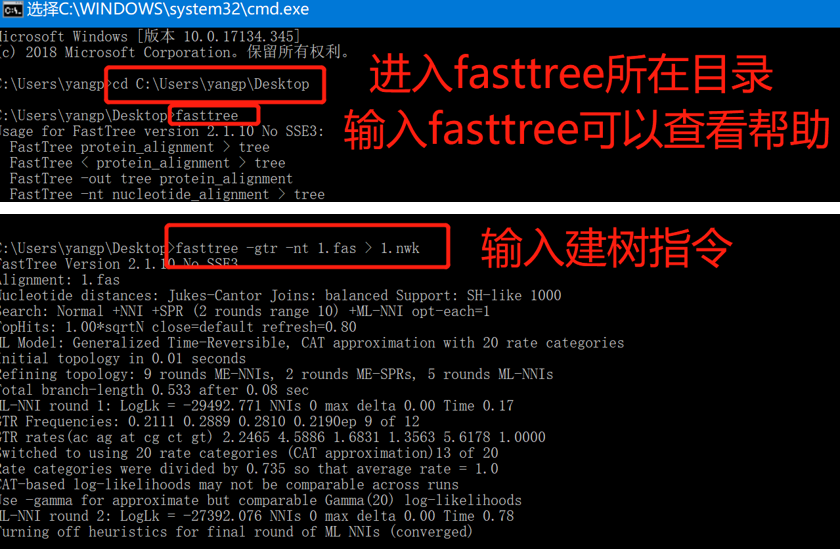
3.1 cmd 切换至FastTree.exe所在目录
# 本文演示时的exe文件存放于桌面,所以路径如下,根基实际情况修改路径
cd C:\Users\yangp\Desktop
# 此时可以输入下面指令获得软件的帮助文档
FastTree.exe --help
Tips:推荐使用 cmdhere 来直接进入 FastTree 所在的目录。
3.2 建树
本文演示时的 exe 文件存放于桌面,根据实际情况修改路径。
## 快速使用——对核酸序列进行建树
FastTree -gtr -nt nucleotides-seq.fas > tree.nwk
# -gtr 使用gtr模型进行建树,默认核酸使用Jukes-Cantor模型
# -nt 指定核酸比对,默认蛋白比对
## 具体示例说明
# 1.对比对后的蛋白序列使用 JTT+CAT 建树可以输入以下指令
FastTree < alignment_file > tree_file
FastTree alignment.file > tree_file
# 添加-wag 表示使用 WAG+CAT 模型进行建树
# 添加-lg 表示使用 LG+CAT 模型进行建树
# 2.对比对后的核酸序列使用 GTR+CAT 建树可以输入以下指令
FastTree -gtr -nt < alignment.file > tree_file
FastTree -gtr -nt alignment_file > tree_file
# If you do not specify -gtr, then FastTree will use the Jukes-Cantor + CAT model instead
# 如果去掉 -gtr 选项,表示使用Jukes-Cantor + CAT模型进行建树
注意:
- .fas 和 .nwk 是文件名,需要对应
- nucleotides-seq.fas需为已比对且修剪好的序列,不只是fas格式文件,faste一类格式的文件都可以
- 建议将FastTree.exe和Alignment-seq.fas放在同一个文件夹内方便管理
- tree.nwk为输出文件,名字可自行修改,格式一般为nwk,默认存放在FastTree.exe同一个目录下
- nwk的文件官方推荐比较小的树用MEGA查看,比较大的数用Archaeopteryx查看,非常大的16S trees用Arb查看
- GTR(general time reversible model) 和 Jukes-Cantor都是比较常用的一种建树算法
3.3 示例
使用一个叫1.fas的比对好的核酸序列文件进行建树,输出一个叫1.nwk的树文件的全过程截图。