
SparK: NGS数据可视化
SparK 是一款基于 Python 来展示基因覆盖度的脚本,而且输出的是 svg 格式的矢量图。在展示 ChIP-seq 结果还是比较方便的。
1.安装
SparK 是一个下载即用的 Python 脚本,推荐使用 Python3 运行,同时需安装 numpy 库。
2.举例说明
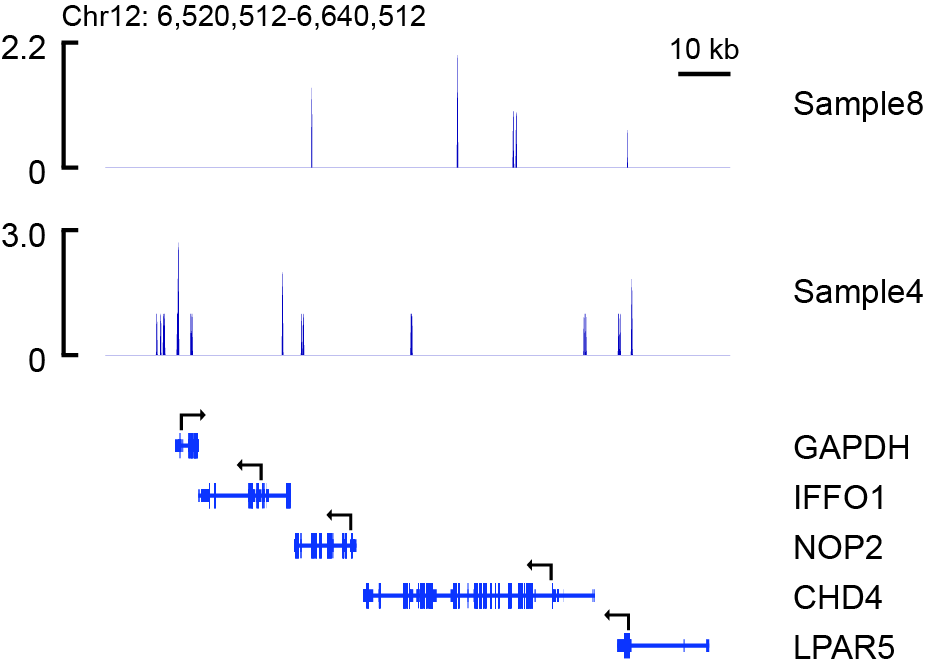
上图代码如下:
python SparK.py \
-pr chr12:6520512-6640512 \ # 作图区域
-cf ./data/Sample8.sorted_dupm.bdg ./data/Sample4.sorted_dupm.bdg \ # 对照组bdg文件,空格区分多个文件
-gtf /home/devdata/Genome/fasta_gtf/Homo_sapiens.GRCh38.93.gtf \ # gtf文件
-gl Sample8 Sample4 \ # 分组标签,顺序与 -cf 对应,空格区分多个文件
-dg GAPDH IFFO1 NOP2 CHD4 LPAR5 \ # 选择展示的基因,若无该参数,展示区域内所有基因
-o TEST1 # 输出文件名
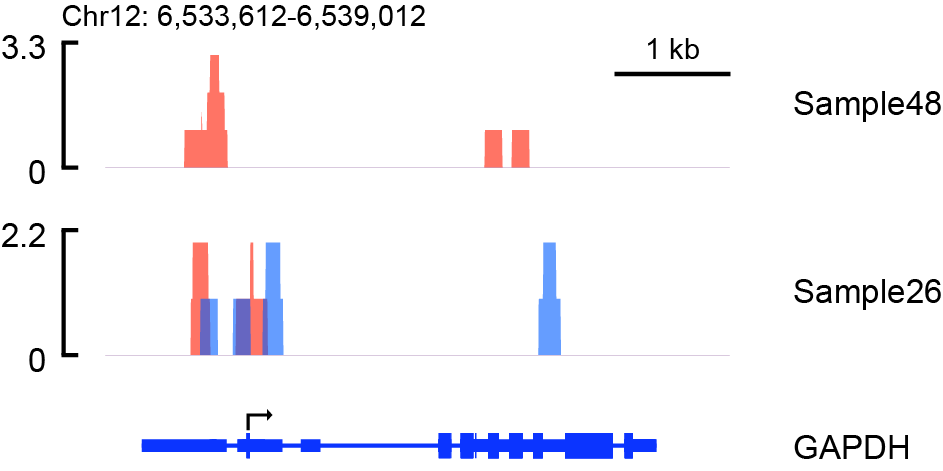
上图代码如下:
python SparK.py \
-pr chr12:6533612-6539012 \
-tf ./data/Sample8.sorted_dupm.bdg ./data/Sample2.sorted_dupm.bdg \ # 处理组bdg文件,空格区分多个文件
-cf ./data/Sample4.sorted_dupm.bdg ./data/Sample6.sorted_dupm.bdg \
-tg 1 2 \ # 处理组分组信息,空格区分多个分组信息
-cg 1 2 \ # 对照组分组信息,空格区分多个分组信息
-gl Sample48 Sample26 \
-gtf /home/devdata/Genome/fasta_gtf/Homo_sapiens.GRCh38.93.gtf \
-dg GAPDH \
-o TEST2
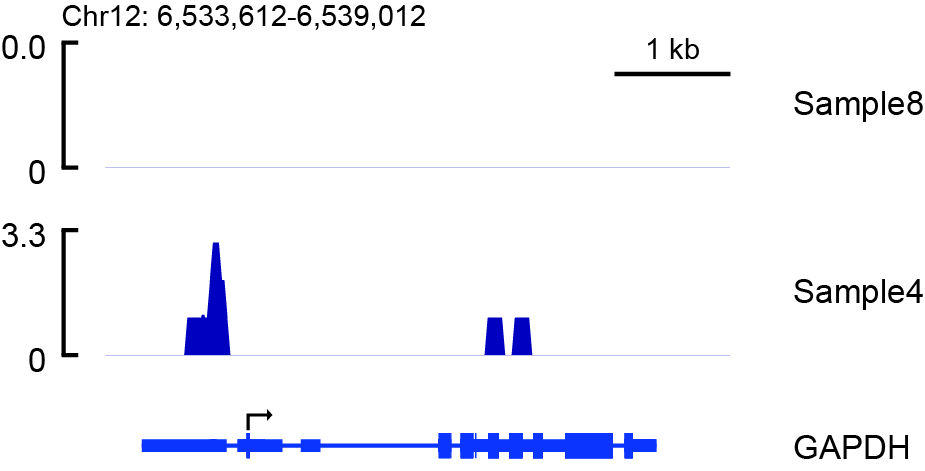
上图代码如下:
python SparK.py \
-pr chr12:6533612-6539012 \
-cf ./data/Sample8.sorted_dupm.bdg ./data/Sample4.sorted_dupm.bdg \
-gtf /home/devdata/Genome/fasta_gtf/Homo_sapiens.GRCh38.93.gtf \
-gl Sample8 Sample4 \
-dg GAPDH \
-sm 10 \ # 平滑轨迹,数字为窗口大小
-o TEST3
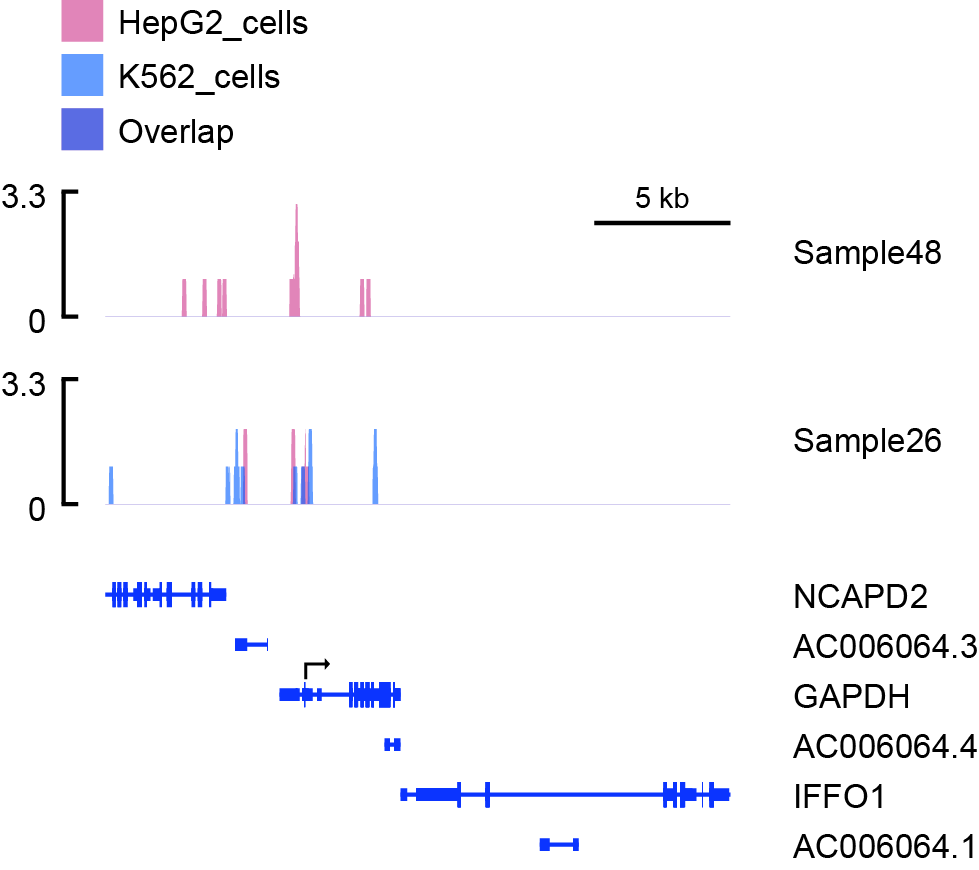
上图代码如下:
python SparK.py \
-pr chr12:6527512-6550512 \
-tf ./data/Sample8.sorted_dupm.bdg ./data/Sample2.sorted_dupm.bdg \
-cf ./data/Sample4.sorted_dupm.bdg ./data/Sample6.sorted_dupm.bdg \
-tg 1 2 \
-cg 1 2 \
-gl Sample48 Sample26 \
-l HepG2_cells K562_cells \
-gtf /home/devdata/Genome/fasta_gtf/Homo_sapiens.GRCh38.93.gtf \
-ps averages \ # Set to "averages" if replicates of control and treatment tracks should be averaged for all groups.
-f CE358B 005CFF \ # 填充色
-gs yes \ # y轴
-dc no \ # 染色体位置,top_left (默认), top_right, bottom_left, bottom_right 和 no
-o TEST4
以上4个例子仅展示了部分参数,还有更多的参数详见软件的 help 文档,若没有相应参数可以修改所需修改的部分,也可以直接用 AI 打开 svg 的输出文件进行更加个性化的编辑。
3.制作 bdg 文件
以上四个示例的输入均是 bdg 文件,并不是常见的 bam、bed 或 bw 文件,所以作者提供了两种转换为 bdg 文件的思路。
3.1 bamCoverage
可以将 bam 文件转换为 bdg 文件,注意必须添加 -bs 1 参数,该参数是将 bin 的大小设置成 1,所以不难想到 bdg 文件里面存储的是每个碱基对应的测序深度。
bamCoverage -b bamfile.bam -o outputfilename.bdg -bs 1 -of bedgraph
3.2 bigWigToBedGraph
需要注意的是 ChIP-seq 的数据不能使用上面方法转换 bdg 文件!!! 需使用 bigWigToBedGraph 将 ChIP-seq 流程中产生的 bw 文件转换成 bgd 文件。
bigWigToBedGraph 也是一个下载即用的工具,下载地址如下。由于功能单一,所以使用也比较简单,使用之前需添加 x 的执行权限(指令:chmod +x bedGraphToBigWig)。
下载地址:http://hgdownload.soe.ucsc.edu/admin/exe/linux.x86_64/
### kent source version 402 ###
bigWigToBedGraph - Convert from bigWig to bedGraph format.
usage:
bigWigToBedGraph in.bigWig out.bedGraph
options:
-chrom=chr1 - if set restrict output to given chromosome
-start=N - if set, restrict output to only that over start
-end=N - if set, restict output to only that under end
-udcDir=/dir/to/cache - place to put cache for remote bigBed/bigWigs
参考资料:
1.SparK - Publication quality NGS data plotting (Version 2.6.2)